In this post, I am going to discuss the genetic disease Phenylketonuria (PKU).
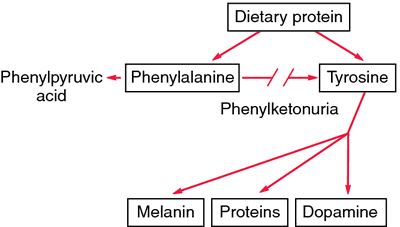
PKU occurs when a genetic mutation is inherited, disrupting the function of the PAH metabolic enzyme. In a functional PAH enzyme, it catalyzes the phenylalanine amino acid into another amino acid: tyrosine. With a defective PAH enzyme, the reaction needed to break down phenylalanine and produce tyrosine does not occur.
http://img.tfd.com/mk/P/X2604-P-23.png
Due to the build up and excess amounts of phenylalanine, multiple symptoms can occur, such as epilepsy, brain retardation, and a musty smell to the skin can occur. It is also possible for a child to have a head size below average expectation as a symptom of PKU. As a result of lack of tyrosine, the most identifiable symptoms lie in appearance. These symptoms include lighter qualities in skin and hair, and often cause eyes to be blue.
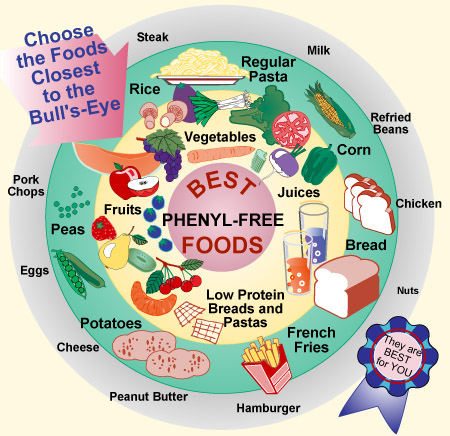
PKU is not a very common disorder, only about 1 in every 15,000 infants are diagnosed with the disease. Though the odds are not in the favor of a to be child to be born with PKU, it is very important that it be identified at an early age. In fact, the common procedure in the United States requires babies to be tested for the disorder immediately after they are born. The reason behind the urgency of testing is an attempt to prevent the build up of phenylalanine and provide treatment for the patient. Treatment is obtained through a low protein diet and the avoidance of eggs, meat, and dairy. Below are a few examples of how PKU patients organize foods into an acceptable dietary plan. The goal is to "hit the bull's eye" and consume phenyl-free foods.

http://www.drkarencann.com/wp-content/uploads/2010/11/Food-Target-Graphic-.jpg
http://depts.washington.edu/pku/images/food_bullseye.jpg
For more information on PKU, please visit:
No comments:
Post a Comment